




















MOLCAS manual: Next: 8.45 seward Up: 8. Programs Previous: 8.43 rpa
|
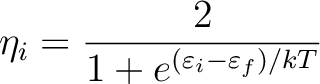 |
(8.12) |
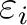 is the orbital energy of orbital i and
is the orbital energy of orbital i and
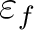 is the Fermi energy.
In this ``Fermi aufbau'' procedure
the temperature is slowly lowered until it reaches a minimum
value and then kept constant until
a stable closed shell configuration is determined.
Then normal SCF iterations will be performed with the selected
closed shell configuration.
For systems that are not really closed shell systems, for example
diradicals, you might end up in the situation that the program
does not find any stable closed shell configuration.
In that case it will continue to optimize the closed shell
energy functional with partial occupation numbers.
If this is the case, this is probably what you want, and such
orbitals would be ideal as starting orbitals for an MCSCF
calculation.
is the Fermi energy.
In this ``Fermi aufbau'' procedure
the temperature is slowly lowered until it reaches a minimum
value and then kept constant until
a stable closed shell configuration is determined.
Then normal SCF iterations will be performed with the selected
closed shell configuration.
For systems that are not really closed shell systems, for example
diradicals, you might end up in the situation that the program
does not find any stable closed shell configuration.
In that case it will continue to optimize the closed shell
energy functional with partial occupation numbers.
If this is the case, this is probably what you want, and such
orbitals would be ideal as starting orbitals for an MCSCF
calculation.
The initial orbital guess is either obtained by diagonalizing the bare nuclei Hamiltonian, from an initial guess produced by the module Guessorb or from orbitals of a previous Hartree-Fock SCF calculation. These starting orbitals are automatically located in the order
- SCF orbitals from a previous calculation located in the RUNFILE
- SCF orbitals from a previous calculation located in a formatted orbitals file, INPORB.
- initial guess orbitals from module Guessorb located in the RUNFILE and
By default SCF behaves in different ways depending on what kind of start orbitals are found according to
- No start orbitals are found. In this case the core hamiltonian is diagonalized and these orbitals are used as start. The ``Fermi aufbau'' procedure is used until a stable configuration is found.
- Start orbitals from Guessorb are found. In this case the HOMO LUMO gap is analyzed and if it is small the ``Fermi aufbau'' procedure is used until a stable configuration is found. Otherwise the configuration suggested by Guessorb is used.
- Start orbitals from a previous SCF calculation is found. The configuration from the previous SCF calculation is used, unless some problem is detected such as partial occupation numbers from an unconverged calculation. In the latter case ``Fermi aufbau'' is used.
- Start orbitals from an INPORB is in the same way as for start orbitals from an SCF calculation, see last point.
One of the main objects of the SCF program in the context of the
MOLCAS program system is to generate starting
orbitals for subsequent MCSCF calculations.
Two options are available to
improve the canonical Hartree-Fock orbitals in this respect.
(i) It is possible to specify pseudo occupation numbers that are neither
zero nor two, thus simulating to some extent an open shell system. The
resulting wavefunction does not have any physical meaning, but will
provide better starting orbitals for open shell systems.
(ii) Usually, the lowest virtual canonical Hartree-Fock orbitals are
too diffuse as correlating orbitals in an MCSCF calculation.
If the keyword IVO is encountered in the input stream, the
SCF program will diagonalize the core Hamiltonian matrix within
the virtual space and write the resulting more compact eigenvectors to the
SCFORB and RUNFILE files,
rather than the virtual eigenvectors of the Fock
matrix. It should be noted, that this option must never be used, if the
SCF wave function itself is used subsequently as a reference
function: no MP2 or coupled cluster calculations after an
SCF run with IVO !
A further method to generate starting orbitals for MCSCF calculations is
to perform an SCF calculation for a slightly positively charged moiety.
8.44.2 Dependencies
The SCF program requires the one-electron integral file
ONEINT and the communications file RUNFILE,
which contains among others the
basis set specifications processed by SEWARD. For conventional
(not integral-direct) runs the two-electron integral file ORDINT
is required as well. All these files are generated by a preceding
SEWARD run.
8.44.3 Files
Below is a list of the files that are used/created by the program SCF.
8.44.3.1 Input files
SCF will use the following input files: ONEINT, ORDINT,RUNFILE, INPORB (for more information see![[*]](crossref.png) ).
).
8.44.3.2 Output files
| File | Contents |
| SCFORB | SCF orbital output file. Contains the canonical Hartree-Fock orbitals for closed shell calculations. If the IVO option was specified, the virtual orbitals instead are those that diagonalize the bare nuclei Hamiltonian within that subspace. |
| UHFORB | Contains the canonical Hartree-Fock orbitals for open shell calculations. |
| UNAORB | This file is produced if you make a UHF calculation and it contain natural orbitals. |
| MD_SCF | Molden input file for molecular orbital analysis. |
8.44.4 Input
Below follows a description of the input to SCF.
The input for each module is preceded by its name like:
&SCF
Argument(s) to a keyword, either individual or composed by several entries, can be placed in a separated line or in the same line separated by a semicolon. If in the same line, the first argument requires an equal sign after the name of the keyword.
8.44.4.1 Basic general keywords
Below is a list of keywords that should cover the needs of most users.| Keyword | Meaning | ||||||||||
| TITLe | One line for the title | ||||||||||
| UHF | Use this keyword to run Unrestricted Hartree-Fock code. Note that current implementation of UHF code has some restrictions, and not all features of SCF program are supported. | ||||||||||
| ZSPIN | Use this keyword to specify the difference in the number of 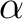 and and 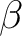 electrons in the system. The default is 0 or 1 depending on if there is an even
or odd number of electrons.
Any value different from 0 requires the UHF keyword.
This keyword is not needed when you specify the number of electrons with
the keyword OCCUpied.
electrons in the system. The default is 0 or 1 depending on if there is an even
or odd number of electrons.
Any value different from 0 requires the UHF keyword.
This keyword is not needed when you specify the number of electrons with
the keyword OCCUpied.
| ||||||||||
| SPIN | Alternative way of specifying the electronic spin of the system. The keyword is followed by an integer giving the value of spin multiplicity (2S+1). Default is 1 (singlet) or 2 (doublet) depending on if there is an even or odd number of electrons. Any value different from 1 requires the UHF keyword. | ||||||||||
| KSDFT | Use this keyword to do density functional theory calculations. This keyword should be followed by functional keyword: BLYP, B3LYP, B3LYP5, HFB, HFS, LDA, LDA5, LSDA, LSDA5, SVWN, SVWN5, TLYP, PBE, PBE0, M06, M062X, M06HF, M06L. Example: KSDFT=B3LYP | ||||||||||
| DFCF | Use this optional keyword to scale the exchange terms and/or correlation terms of the density functional requested with KSDFT. This keyword should be followed by the scaling factor for the exchange terms and the scaling factor for the correlation terms, separated by a space. If the values are 1.0 (default), then the original density functional is used. For an HLE-type functional, use 1.25 (for exchange) and 0.5 (for correlation). Example: DFCF=1.25 0.5 | ||||||||||
| CHARge | Use this keyword to set the number of electrons in the system.
This number is defined by giving the net charge of the system.
If this keyword is not specified, the molecule is assumed to
have net charge zero.
The input is given as
Charge=n where n is the charge of the system.
| ||||||||||
| OCCUpied | Use this keyword to set the number of electrons in the system.
This number is defined by giving the number of electron pairs
per irreducible representation of the subgroup of D2h used
in the calculation.
You can use one and only one of the keywords,
CHARge and
OCCUpied for this purpose.
If neither of these keywords are specified
CHARge is assumed with a net charge of zero.
It should be noted that the ``fermi aufbau''
procedure is not used when you specify this keyword.
The input for one of the point groups D2, C2h or C2v
is given as
OCCUpied= n1 n2 n3 n4 where n1 is the number of electron pairs (occupied orbitals)
in the first irreducible representation, etc.
If UHF keyword was specified, occupation numbers must be specified in two lines: for alpha and beta spins | ||||||||||
| FERMi | Use this keyword to specify that you want to use the ``Fermi aufbau''
procedure for the first few iterations to ensure convergence.
The orbitals will be partially populated according to a Fermi
population.
The input is gives as
Fermi= m where m is the temperature parameter according to
| ||||||||||
| CHOLesky | SCF will use Cholesky (or RI/DF) representation of the two-electron integrals to compute the corresponding contributions to the Fock or KS matrices. The default (LK) algorithm is used. The configuration may be tailored using the ChoInput section. Default is to not use Cholesky unless the Cholesky (or RI/DF) representation of the two-electron integrals has been produced by SEWARD. | ||||||||||
| CHOInput | This marks the start of an input section for modifying
the default settings of the Cholesky SCF.
Below follows a description of the associated options.
The options may be given in any order,
and they are all optional except for
ENDChoinput which marks the end of the CHOInput section.
| ||||||||||
| CONStraints | Performs a Constrained (Natural Orbitals) SCF calculation, available only in combination with Cholesky or RI integral representation.
An example of input for the keyword CONS is the following:
CONStraints 2 3 1 -1 1 1 1 ADDCorrelation pbe SAVErage The keyword CONS has two compulsory arguments: the number of constrained NOs (in each irrep) to be used in the CNO-SCF calculation, followed by one line per irrep specifying the spin configuration of the so-called (+) wavelet (-1 –> beta, 1 –> alpha) The OPTIONAL keyword ADDC is used to include a correlation energy correction through a DFT functional specified as argument (LDA, LDA5, PBE and BLYP available at the moment) The OPTIONAL keyword SAVE forces the program to use spin-averaged wavelets. | ||||||||||
| OFEMbedding | Performs a Orbital-Free Embedding (OFE)SCF calculation, available only in combination with Cholesky or RI integral representation.
The runfile of the environment subsystem renamed AUXRFIL is required.
An example of input for the keyword OFEM is the following:
OFEMbedding ldtf/pbe dFMD 1.0 1.0d2 FTHAw 1.0d-4 The keyword OFEM requires the specification of two functionals in the form fun1/fun2, where fun1 is the functional used for the Kinetic Energy (available functionals: Thomas-Fermi, with acronym LDTF, and the NDSD functional), and where fun2 is the xc-functional (LDA, LDA5, PBE and BLYP available at the moment). The OPTIONAL keyword dFMD has two arguments: first, the fraction of correlation potential to be added to the OFE potential (zero for KSDFT and one for HF); second, the exponential decay factor for this correction (used in PES calculations). The OPTIONAL keyword FTHA is used in a freeze-and-thaw cycle (EMIL Do While) to specify the (subsystems) energy convergence threshold. | ||||||||||
| ITERations | Specifies the maximum number of iterations. The default is 400 which
is also the largest number you can specify.
| ||||||||||
| CORE | The starting vectors are obtained from a diagonalization of the core Hamiltonian. | ||||||||||
| LUMORB | The starting vectors are taken from a previous SCFORB file called INPORB. | ||||||||||
| FILEORB | The starting vectors are taken from a previous SCFORB file, specified by user. | ||||||||||
| GSSRunfile | The starting vectors are taken from the orbitals produced by Guessorb. | ||||||||||
| HLGAp | This keyword is used to make the program level shift the virtual orbitals in such a way that the HOMO LUMO gap is at least the value specified on the next line. This will help convergence in difficult cases but may lead to that it converges to an excited configuration. A suitable value is 0.2. |
8.44.4.2 Advanced general keywords
| Keyword | Meaning |
| SCRAmble | This keyword will make the start orbitals slightly scrambled,
accomplished by making a few small random orbital rotations.
How much the orbitals are scrambled is determined by the
parameter read on the next entry. A reasonable choice for
this parameter is 0.2 which correspond to maximum rotation angle
of 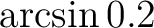 .
Using this keyword may be useful for UHF calculations with
same number of and electrons that are not
closed shell cases. .
Using this keyword may be useful for UHF calculations with
same number of and electrons that are not
closed shell cases.
|
| ORBItals | Specifies the number of orbitals in the subspace of the full orbital space defined by the basis set, in which the SCF energy functional is optimized. The size of this subspace is given for each of the irreducible representations of the subgroup of D2h. If this keyword is not specified when starting orbitals are read, the full orbital space is assumed. The keyword takes as argument nIrrep (# of irreps) integers. Note that this keyword is only meaningful when the SCF program is fed with input orbitals (cf. LUMORB). |
| FROZen | Specifies the number of orbitals not optimized during iterative procedure. The size of this subspace is given for each of the irreducible representations of the subgroup of D2h. If this keyword is not specified the number of frozen orbitals is set to zero for each irreducible representation. If the starting vectors are obtained from a diagonalization of the bare nuclei Hamiltonian the atomic orbitals with the lowest one-electron energy are frozen. If molecular orbitals are read from INPORB the frozen orbitals are those that are read in first in each symmetry. The keyword takes as argument nIrrep (# of irreps) integers. |
| OVLDelete | Specifies the threshold for deleting near linear dependence in the basis set. The eigenvectors of the overlap matrix with eigenvalues less than that threshold are removed from the orbital subspace, and do not participate in the optimization procedure. The default value is 1.0d-5. The keyword takes as argument a (double precision) floating point number. Note that the SCFORB file will contain the deleted orbitals as a complementary set to the actual SCF orbitals! In future use of this orbital file the complementary set should always be deleted from use. |
| PRORbitals | Specifies which orbitals are to be printed in the log file (standard output).
The keyword takes as argument two integers.
The possible values are:
Second (optional) argument specifies a format:
|
| PRLScf | Specifies the general print level of the calculation. An integer has to be supplied as argument. The default value, 1, is recommended for production calculations. |
| ROBU | Robust LDF integral representation (non-hybrid KS-DFT only). Requires Local Density Fitting (LDF) in SEWARD. This is the default for LDF. |
| NR-2 | Nonrobust LDF integral representation with 2-index integrals only (non-hybrid KS-DFT only). Requires Local Density Fitting (LDF) in SEWARD. Default is robust integral representation. |
| NR-3 | Nonrobust LDF integral representation with 3-index integrals only (non-hybrid KS-DFT only). Requires Local Density Fitting (LDF) in SEWARD. Default is robust integral representation. |
| XIDI | Use exact integral diagonal blocks with LDF. Reduces the risk of negative eigenvalues of the approximate integral matrix. Default is to not use exact integral diagonal blocks. |
| THREsholds | Specifies convergence thresholds. Four individual thresholds are specified as arguments, which have to be fulfilled simultaneously to reach convergence: EThr, DThr and FThr specify the maximum permissible difference in energy, density matrix elements and Fock matrix elements, respectively, in the last two iterations. The DltNTh finally specifies the norm of the orbital displacement vector used for the orbital rotations in the second-order/C2-DIIS procedure. The corresponding values are read in the order given above. The default values are 1.0d-9, 1.0d-4, 1.5d-4, and 0.2d-4, respectively. Note that these thresholds automatically define the threshold used in the direct Fock matrix construction to estimate individual contributions to the Fock matrix such that the computed energy will have an accuracy that is better than the convergence threshold. |
| NODIis | Disable the DIIS convergence acceleration procedure. |
| DIISthr | Set the threshold on the change in density, at which the DIIS procedure is turned on. The keyword takes as argument a (double precision) floating point number. The default value is 0.15. |
| QNRThr | Set the threshold on the change in density, at which the
second-order/C2-DIIS
procedure kicks in.
The keyword takes as argument a (double precision) floating point number.
The default value is 0.15.
Note: the change in density has to drop under both the DIISthr and the QNRThr threshold, for the second-order/C2-DIIS to be activated. If the latter is set to zero the older first order C2-DIIS procedure will be used instead. |
| C1DIis | Use C1-DIIS for convergence acceleration rather than C2-DIIS which is the default (not recommended). |
| NODAmp | Disable the Damping convergence acceleration procedure. |
| OCCNumbers | Gives the option to specify occupation numbers other than 0 and 2. This can be useful for generating starting orbitals for open shell cases. It should be noted however, that it is still the closed shell SCF energy functional that is optimized, thus yielding unphysical energies. Occupation numbers have to be provided for all occupied orbitals. In the case of UHF calculation occupation numbers should be specified on two different entries: for alpha and beta spin. |
| IVO | Specifies that the virtual orbitals are to be improved for subsequent MCSCF calculations. The core Hamiltonian is diagonalized within the virtual orbital subspace, thus yielding as compact orbitals as possible with the constraint that they have to be orthogonal to the occupied orbitals. Note that this option must not be used whenever the Hartree-Fock wavefunction itself is used as a reference in a subsequent calculation. |
| NOMInimization | Program will use density differences
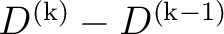 rather than minimized differences.
rather than minimized differences.
|
| ONEGrid | Disable use of a smaller intermediate grid in the integration of the DFT functional during the first SCF iterations. |
| RFPErt | This keyword will add a constant reaction field perturbation to the bare nuclei hamiltonian. The perturbation is read from RUNOLD (if not present defaults to RUNFILE) and is the latest self consistent perturbation generated by one of the programs SCF or RASSCF. |
| STAT | This keyword will add an addition print outs with statistic information.
|
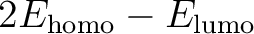 are printed.
are printed.
For calculations of a molecule in a reaction field see section
of the present manual and section of the examples manual.
8.44.4.2.1 DFT functionals:
Below is listed the keywords for the DFT functionals currently implemented in the package.| Keyword | Meaning | ||
| LSDA, LDA, SVWN | Vosko, Wilk, and Nusair 1980 correlation functional fitting the RPA solution to the uniform electron gas [100] (functional III in the paper). | ||
| LSDA5, LDA5, SVWN5 | Functional V from the VWN80 paper [100] which fits the Ceperley-Alder solution to the uniform electron gas. | ||
| HFB | Becke's 1988 exchange functional which includes the Slater exchange along with corrections involving the gradient of the density [101]. | ||
| HFS | 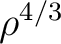 with the theoretical coefficient of 2/3 also known as Local Spin Density exchange
[102,103,104]. with the theoretical coefficient of 2/3 also known as Local Spin Density exchange
[102,103,104].
| ||
| HFB86 | Becke's 1986 two-parameter exchange functional which includes the Slater exchange along with corrections involving the gradient of the density [105,106]. | ||
| HFO | Handy's stand-alone OPTX exchange functional [107] | ||
| BLYP | Becke's 1988 exchange functional which includes the Slater exchange along with corrections involving the gradient of the density [101]. Correlation functional of Lee, Yang, and Parr, which includes both local and non-local terms [108,109]. | ||
| BPBE | Becke's 1988 exchange functional which includes the Slater exchange along with corrections involving the gradient of the density [101] , combined with the GGA correlation functional by Perdew, Burke and Ernzerhof [110] | ||
| B3LYP | Becke's 3 parameter functional [111] with the form
| ||
| B3LYP5 | Becke's 3 parameter functional [111] with the form
| ||
| B2PLYP | Grimme's double-hybrid density functional [112] based on Becke;s 1988 exchange and LYP correlation GGA functionals with the form
| ||
| B86LYP | Becke's 1986 exchange [105,106] functional combined with the LYP correlation [108,109] | ||
| BWig | Becke's 1988 GGA exchange functional combined with the local Wigner correlation functional [113] | ||
| GLYP | Gill's 1996 GGA exchange functional [114] combined with the combined with the LYP correlation [108,109] | ||
| OLYP | Handy's OPTX exchange functional [107] combined with the LYP correlation [108,109] | ||
| OPBE | Handy's OPTX exchange functional [107] combined with the PBE correlation[110] | ||
| O3LYP | A hybrid density functional based on the OPTX exchange [115] , with the form
| ||
| KT3 | The exchange-correlation functional by Keal and Tozer, 2004 [116,117] | ||
| TLYP |
where the non-local correlation functional is the LYP functional | ||
| PBE | The Perdew, Burke, Ernzerhof GGA functional 1996 [110]. | ||
| PBE0 | The Perdew, Ernzerhof, Burke non-empirical hybrid functional 1996 [118]. | ||
| PBEsol | The Perdew et al. 2008 modification of PBE for solids | ||
| RGE2 | The regularized gradient approximation (RGE2 )exchange functional by Ruzsinszky, Csonka, and Scuseria, 2009 that contains higher-power s terms in the exchange functional, as compared to the PBEsol. It is coupled with the PBEsol correlation [119] | ||
| PTCA | The correlation functional by Tognetti, Cortona, and Adamo combined with the PBE exchange [120] | ||
| SSB | The exchange functional SSB-sw by Swart, Sola, and Bickelhaupt, 2008 [121] that switches between the OPTX exchange for small values of the reduced density gradient and the PBE exchange for the large ones. It is combined with the PBE correlation functional. | ||
| M06 | The M06 functional of the Minnesota 2006 class of functionals by Zhao and Truhlar [122,123,124,125] | ||
| M06L | The M06-L functional of the Minnesota 2006 class of functionals by Zhao and Truhlar [122,123,124,125] | ||
| M06HF | The M06-HF functional of the Minnesota 2006 class of functionals by Zhao and Truhlar [122,123,124,125] | ||
| M062X | The M06-2X functional of the Minnesota 2006 class of functionals by Zhao and Truhlar [122,123,124,125] |
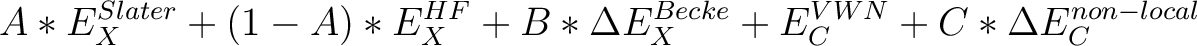
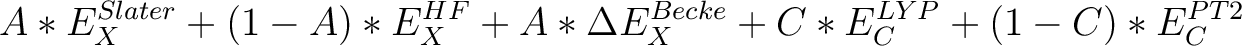
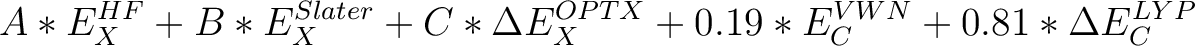
8.44.4.3 Keywords for direct calculations
Note again that the threshold for contributions to the Fock matrix depends on the convergence thresholds mentioned above. The choice between the conventional and direct SCF methods is based on the presence of a two-electron integral file (file ORDINT). The keyword Direct in the SEWARD input controls that no two-electron integral file is to be generated and that integral direct algorithms can be used in subsequent modules. Thus, the choice between conventional and direct SCF is done already in the input for the integral program SEWARD. The direct (or semi-direct) path will be taken whenever there are no two-electron integrals available.
| Keyword | Meaning |
| CONVentional | This option will override the automatic choice between the conventional and the direct SCF algorithm such that the conventional method will be executed regardless of the status of the ORDINT file. |
| DISK | This option enables/disables the semi-direct algorithm. It requires two arguments which specifies the max Mbyte of integrals that are written on disk during the first iteration (and retrieved later in subsequent iterations) and the size of the corresponding I/O buffer in kbyte. The default values are 2000 MByte and 512 kByte. In case the specified disk space is zero and the I/O buffer is different from zero it will default to a semi-direct SCF with in-core storage of the integrals. The size of the memory for integrals storage is the size of the I/O buffer. If the size of the disk is non-zero and the I/O buffer size is zero the latter will be reset to the default value. |
| THIZe | This option specifies a threshold for two-electron integrals. Only integrals above this threshold (but not necessarily all of those) are kept on disk for the semi-direct algorithm. The keyword takes as argument a (double precision) floating point number. |
| SIMPle | If this option is specified, only a simple prescreening scheme, based solely on the estimated two-electron integral value will be employed (no density involved). |
8.44.4.4 Limitations
The limitations/MODULE on the number of basis functions are the same as specified for SEWARD.
8.44.4.5 Input examples
First we have the bare minimum of input. This will work well for almost all systems containing an even number of electrons.
&SCF
The next example is almost as simple. Here we have an open shell case, i.e. you have an odd number of electrons in the neutral system and you need to generate starting orbitals for RASSCF. In this case we recommend that you perform a calculation on the cation with the input below.
&SCF; Charge= 1
The next example explains how to run UHF code for a nitrogen atom:
&SCF; UHF; ZSPIN=3
The next example is a bit more elaborate and show how to use
a few of the keywords. The system is water that have the
electron configuration
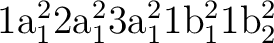 .
.
&SCF; Title= Water molecule. Experimental equilibrium geometry. The symmetries are a1, b2, b1 and a2.
Occupied= 3 1 1 0
Threshold= 0.5D-9 0.5D-6 0.5D-6 0.5D-5
* semi-direct algorithm writing max 128k words (1MByte) to disk
* the size of the I/O buffer by default (512 kByte)
Disk= 1 0
Ivo
Next: 8.45 seward Up: 8. Programs Previous: 8.43 rpa

