




















MOLCAS manual: Next: 8.18 genano Up: 8. Programs Previous: 8.16 ffpt
|
| Keyword | Meaning |
| TITLE | The keyword followed by a title.
|
| BASDIR | The keyword allows to set up an extra location for basis set files.
The value can be either an absolute path (started from /) or relative to
submit directory, e.g. BASDIR=.
In order to use a local copy of a basis set file with name FOO - place
this file into directory specified in BASDIR
|
| BASLIB | The keyword followed by the absolute path to the basis set library directory. The default
is the $MOLCAS/basis_library directory. Note that this directory must also be host to
local copies of the .tbl files.
|
| RTRN | Max number of atoms for which bond lengths, angles and dihedral
angles are listed, and
the radius defining the maximum length of a bond follows on
the next line. The latter is used as a threshold when printing out
angles and dihedral angles. The length can be followed by
Bohr or
Angstrom which indicates the unit in which the length
was specified, the default is
Bohr.
The default values are 15 and 3.0 au.
|
| ISOTopes | Specify isotopic substitutions or atomic masses. By default, the mass of the most
abundant or stable isotope is used for each atom. With this keyword different
isotopes or arbitrary masses can be chosen. The keyword is followed by the number
n of isotopic specifications, and then by n lines. Each of these
lines should contain the symmetry-unique index of the atom for which the default
mass is to be modified and either the mass number of the desired isotope (tabulated
masses for most known isotopes are available in the code, use 0 for the default
isotope) or the desired mass in dalton, in the latter case the keyword Dalton
should follow. Note that all atoms belonging to the same ``center type'' must have the
same mass. This usually means all atoms of a given element with the same basis set.
If more fine-grained specifications are wanted, additional center types must be
created by using several BASIs Set blocks for the same element (native
input) or by using labels (XYZ input).
|
8.17.1.2 Molecular structure: coordinates, symmetry and basis sets
There are three different ways to specify the molecular structure, symmetry and the basis sets in Gateway:- the so-called native input (old MOLCAS standard),
- XYZ input and
The three different modes will be described below.
8.17.1.2.1 Native input
If the geometry is specified in a native MOLCAS format, only symmetry inequivalent atoms should be specified. The default units are atomic units. By default, symmetry is not used in the calculation.
| Keyword | Meaning |
| SYMMetry | Symmetry specification follows on next line. There may be up to
three different point group generators specified on that line. The
generators of a point group is the minimal set of symmetry operators
which is needed to generate all symmetry
operators of a specific point group. A generator is in the input
represented as a sequence of up to three of the characters x, y, and
z. The order within a given sequence is arbitrary and the generators
can be given in any sequence. Observe that the order of the irreps
is defined by the order of the generators as
( E, g1, g2, g1g2, g3, g1g3, g2g3,
g1g2g3)! Note that E is always assumed and should never
be specified.
Below is listed the possible generators.
|
| BASIs Set | This notes the start of a basis set definition.
The next line always contains a basis set label.
The basis set definition is alway terminated with the "End of Basis" keyword.
For the definitions of basis set labels see the subsequent sections.
Below follows a description of the options associated with the
basis set definition.
|
 4s3p2d., which is an ANO basis set.
If no option is specified
GATEWAY will look for the basis
set in the default basis directory. If an option is specified it
could either be the name of an alternative basis directory or
the wording ``Inline'' which defines
that the basis set will follow in the current input
file. For the format of the
Inline option see the section
`Basis set format'. Observe that the label is arbitrary for this
option and will not be decoded.
The Label card is mandatory.
4s3p2d., which is an ANO basis set.
If no option is specified
GATEWAY will look for the basis
set in the default basis directory. If an option is specified it
could either be the name of an alternative basis directory or
the wording ``Inline'' which defines
that the basis set will follow in the current input
file. For the format of the
Inline option see the section
`Basis set format'. Observe that the label is arbitrary for this
option and will not be decoded.
The Label card is mandatory.
Example of an input in native MOLCAS format:
&GATEWAY
Title
formaldehyde
SYMMETRY
X Y
Basis set
H.STO-3G....
H1 0.000000 0.924258 -1.100293 /Angstrom
End of basis
Basis set
C.STO-3G....
C3 0.000000 0.000000 -0.519589 /Angstrom
End of basis
Basis set
O.STO-3G....
O 0.000000 0.000000 0.664765 /Angstrom
End of basis
End of input
8.17.1.2.2 Z-matrix and XYZ input
Some times it is more convenient to set up information about coordinates in a standard form of Z-matrix or Cartesian coordinates. In this case, the basis set for the atoms should be specified after the XBAS keyword. After that either ZMAT or XYZ should appear to specify the coordinates. Note that coordinates in these formats use ångström as units.
| Keyword | Meaning |
| XBAS | A keyword to specify the basis for atoms. The specification is very similar
to the native format: ATOM.BasisSet. Each new eatom is written at a new line.
The end of the keyword is marked by an 'End of basis' line.
If all atoms have the same basis, e.g. ANO-S-VDZ, it is possible to use this name without element name. In this case there is no need to specify 'End of basis'. Example:
or
|
| ZMAT | Alternative format to give coordinates in terms of bond lengths,
bond angles, and dihedral angles.
Each line of a Z-matrix gives the
internal coordinates for one of the atoms within the molecule with the following
syntax:
Name I bond-length J bond-angle K dihedral-angle Name is the label (atomic symbol + string) for a symmetry distinct center L; I bond-length distance of L from atom I; J bond-angle planar angle between atoms L-I-J; K dihedral-angle dihedral angle between atoms L-I-J-K. Note that the first atom only requires the Name and defines the origin of Cartesian axis. The second atom requires Name I bond-length and it will define the Z axis. The third atom requires Name I bond-length J bond-angle and defines the XZ plane (and implicitly, the Y axis). Only numerical values must be used (no variable names) and ångströms and degrees are assumed as units.
Two types of special atoms are allowed: dummy X atoms and
ghost Z atoms. The former will appear in the calculations,
they have a nuclear charge of 0 and have not electrons and Basis Set.
They will also appear in the definition of internal coordinates in SLAPAF.
The latter are used only within the Z-Matrix definition of the geometry but
they will appear in the final Z-matrix section in SLAPAF.
Both special atoms can be used to define the Cartesian axis and the symmetry elements.
|
| XYZ | The keyword is followed by XYZ formatted file (a reference to a file), or file, inlined into the input. Example:
or
Currently, the XYZ keyword does not operate with symmetry, and the calculation is always performed without symmetry.
|
8.17.1.2.3 Advanced XYZ input
If the geometry is specified in XYZ format, all atoms should be specified. The default units are Ångstroms. By default, maximum possible symmetry is used.'Molcas XYZ' file format is an extension of plain XYZ format.
| First line of this file contains the number of atoms. |
| Second line (a comment line) can contain 'a.u.' or 'bohr' to use atomic units, instead of default Ångstroms. Also this line can contain keyword TRANS, followed by 3 numbers, and/or ROT, followed by 9 numbers (in this case coordinates will be Translated by specified vector, and/or Rotated), and SCALE (or SCALEX, SCALEY, SCALEZ) followed by a scale factor. |
| Remaining lines are used to specify Element and cartesian
coordinates.
Element name might be optionally followed by a Number (e.g. H7),
a Label (separated by _ sign: e.g. |
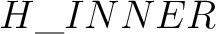 ), or Basis Set (separated by . ,
e.g. H.STO-3G)
), or Basis Set (separated by . ,
e.g. H.STO-3G)
| Keyword | Meaning |
| COORD | The keyword followed on the next line by the name of an HDF5 file (created by any module), or the name of an XYZ file, or inline coordinates in XYZ format. If the file is located in the same directory, where MOLCAS job was submitted there is no need to specify the PATH to this file. The keyword may appear several times. In this case all coordinate files will be concatenated, and considered as individual fragments.
|
| BASIS | The keyword can be used to specify global basis set for all atoms, or for a group of atoms.
The keyword followed by a label of basis set, or by comma separated list of basis sets for
individual atoms.
Note! The basis set definition in XYZ mode does not allow to use inline basis set.
Example:
In this example, the C atom (in the origin) will have the basis set STO-3G and the H atoms 6-31G*. If keyword BASIS never appears in the input, the default basis, ANO-S-MB, will be used.
|
| GROUP | The keyword can be used to specify the symmetry of the molecule. The keyword must be followed by one of:
Limitations: in the current implementation atom labels, and basis sets are ignored during symmetry recognition.
|
If XYZ input has been used in gateway, a file with native MOLCAS input will be produced and stored in working directory under the name findsym.std.
Note that choosing XYZ input you are expecting that the coordinates might be transformed. It can be shown by the following example:
&gateway coord 3 O 0 0 0 H 1.0000001 0 0 H 0 1 0.0000001 *nomove *group=c1
The geometry of the molecule is slightly distorted, but within a threshold it is C2v. Thus by default (keywords nomove and group are not active), the coordinates will be transformed to maintain the highest possible symmetry. If keyword nomove is active, the molecule is not allowed to rotate, and a mirror plane XY is the only symmetry element. Since the third hydrogen atom is slightly out of this plane, it will be corrected. Only activation of the keyword group=C1 will ensure that the geometry is unchanged.
Advanced keywords:
| Keyword | Meaning |
| SYMThreshold | followed by a real number - threshold for symmetry recognition (default is 0.01 Å)
|
| MOVE | allow to translate and rotate molecule in order to find highest possible symmetry. (this is a default for all groups, except of C1) |
| NOMOVE | do not allow to transform coordinates while searching for highest group (default for C1 group)
|
| BSSE | followed by an integer. Indicates which XYZ-file that should be
treated like ghost atoms.
|
| VART | Specifies that the energy should not be considered invariant to translations.
Translational variance is detected automatically, but sometimes it may be useful to enforce it.
|
| VARR | Specifies that the energy should not be considered invariant to rotations.
Rotational variance is detected automatically, but sometimes it may be useful to enforce it.
|
| NUMErical | Forces the calculation of numerical gradients even when analytic gradients are available.
|
| SHAKe | Randomly modifies the initial coordinates of the atoms, maintaining the input (or computed)
symmetry. This can be useful to avoid a geometry optimization converging to a higher-symmetry
saddle point. The maximum displacement in the axes x, y and z is read from the following
real number. This number can be followed by Bohr or Angstrom, which indicates
the unit in which the displacement is specified, the default is Bohr.
|
Example:
&GATEWAY
COORD
water.xyz
BASIS
STO-3G
or, in short EMIL notation:
&GATEWAY
COORD=water.xyz; BASIS=STO-3G
Coordinate file may contain variables, as demonstrated in an example:
>>FILE H2.input
2
scale $DD
H 0.35 0 0
H -0.35 0 0
>>EOF
>> FOREACH DD IN ( 0.9 1.0 1.1 )
&GATEWAY
COORD=$WorkDir/H2.input
BASIS=STO-3G
&SEWARD
&SCF
>>ENDDO
The atom name in XYZ file can contain an orbitrary label (to simplify assigning of different
basis sets). To indicate the label, use _: e.g. 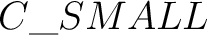 . The same label should be
defined in the basis section:
. The same label should be
defined in the basis section:
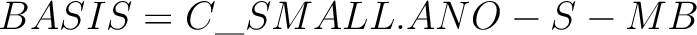 . The basis set label can be also
added into the name of an element:
. The basis set label can be also
added into the name of an element:
COORD
1
O.ANO-S-VDZP 0 0 0
XYZ file can also contain information about point charges. There are three possibilities to setup atomic charges in XYZ file. The main option is to use Q as an element name, and in this case the forth number, the charge, should be specified. Another possibility is to use element names ended with minus sign. In this case, a formal charge for the element will be used. E.g. H-, Li-, Na-, K- defines +1 charge located in the corresponding location. Mg-, Ca- - defines charge +2, Al- – +3, C-, Si- +4, for anions, F-, Cl-, Br-, I- defines -1, O-, S- - -2. Finally, a label at the comment line of XYZ file - CLUSTER followed by an integer number can specify how many atoms are 'real', so the rest will be treated as charges with default values for this element.
8.17.1.3 Constraints
In case of optimizations with constraints these are defined in the GATEWAY input. For a complete description of this keyword see the section![[*]](crossref.png) .
.
| Keyword | Meaning |
| CONStraints | This marks the start of the definition of the constraints which the optimization is subject to. This section is always ended by the keyword End of Constraints. This option can be used in conjunction with any definition of the internal coordinates. |
| NGEXclude | This marks the start of the definition of additional restrictions for numerical differentiation. This section is always ended by the keyword End of NGExclude. The syntax of this section is like that of normal constraints, and the degrees of freedom specified here will be excluded from numerical differentiation (like phantom constraints). If a line containing only ``Invert'' is included inside the section, the definition is reversed and only these degrees of freedom are differentiated. NGEXclude is intended for use with the KEEPOldGradient keyword in ALASKA, and can be combined with CONStraints, which will further reduce the numerical differentiation subspace [20]. Note that the value assigned to the constraints in this section is unused, but a ``Value'' block must still be included. |
8.17.1.4 Explicit auxiliary basis sets
The so-called Resolution of Identity (RI) technique (also called Density Fitting, DF) is implemented in the MOLCAS package. This option involves the use of an auxiliary basis set in the effective computation of the 2-electron integrals. MOLCAS incorporates both the use of conventionally computed, externally provided, auxiliary basis sets (RIJ, RIJK, and RIC types), and on-the-fly generated auxiliary basis sets. The latter are atomic CD (aCD) or the atomic compact CD (acCD) basis sets, based on the Cholesky decomposition method. The externally provided auxiliary basis sets are very compact, since they are tailored for special wave function methods. However, they are not provided for all available valence basis sets. The aCD or acCD RI auxiliary basis sets are a more general option and provides auxiliary basis sets for any wave function model and valence basis set.
| Keyword | Meaning |
| RIJ | Use the RI-J basis in the density fitting (DF) approach to treat the two-electron integrals. Note that the valence basis set must have a supporting auxiliary basis set for this to work. |
| RIJK | Use the RI-JK auxiliary basis in the density fitting (DF) approach to treat the two-electron integrals. Note that the valence basis set must have a supporting auxiliary basis set for this to work. |
| RIC | Use the RI-C auxiliary basis in the density fitting (DF) approach to treat the two-electron integrals. Note that the valence basis set must have a supporting auxiliary basis set for this to work. |
| RICD | Use the aCD or acCD approach [7] to treat the two-electron integrals. This procedure will use an on-the-fly generated auxiliary basis set. |
| CDTHreshold | Threshold for on-the-fly generation of aCD or acCD auxiliary basis sets for RI calculations (default value 1.0d-4). |
| SHAC | Skip high angular combinations à la Turbomole when creating on-the-fly basis sets (default off). |
| KHAC | Keep high angular combinations when creating on-the-fly basis sets (default on). |
| aCD basis | Generate an atomic CD (aCD) auxiliary basis sets (default off). |
| acCD basis | Generate an atomic compact CD (acCD) auxiliary basis sets (default on). |
8.17.1.5 Reaction field calculations
The effect of the solvent on the quantum chemical calculations has been introduced in MOLCAS through the reaction field created by the surrounding environment, represented by a polarizable dielectric continuum outside the boundaries of a cavity containing the solute molecule. MOLCAS supports Self Consistent Reaction Field (SCRF) and Multi Configurational Self Consistent Reaction Field (MCSCRF) calculations within the framework of the SCF and the RASSCF programs. The reaction field, computed in a self-consistent fashion, can be later added as a constant perturbation for the remaining programs, as for example CASPT2.
The purpose of this facility is to incorporate the effect of the environment (a solvent or a solid matrix) on the studied molecule. The utility itself it is not a program, but requires an additional input which has to be provided to the GATEWAY program. Two methods are available for SCRF calculations: one is based on the Kirkwood model, the other is the so called Polarizable Continuum Model (PCM). The reaction field is computed as the response of a dielectric medium polarized by the solute molecule: the solute is placed in a ``cavity'' surrounded by the dielectric. In Kirkwood model the cavity is always spherical, whereas in PCM the cavity is modeled on the actual solute shape.
The possible set of parameters controlled by input are:
- the Kirkwood model,
- the PCM model, and
- one-electron integrals representing Pauli repulsion and external fields.
8.17.1.5.1 The Kirkwood Model
The Kirkwood model is an expansion of the so-called Onsager model where
the surrounding will be characterized by its dielectric
permitivity and a radius describing a spherical cavity,
indicating where the dielectric medium starts.
(Note that all atoms in the studied molecule must be inside the spherical cavity.) The Pauli repulsion
due to the medium can be introduced by use of the spherical well
integrals which are generated by SEWARD.
The charge distribution of the molecule will introduce
an electric field acting on the dielectric medium. This reaction field will interact with the
charge distribution of the molecule. This interaction will manifest itself as
a perturbation to the one-electron Hamiltonian. The perturbation will be
automatically computed in a direct fashion (no multipole integrals are stored on
disk) and added to the one-electron Hamiltonian. Due to the direct way in which
this contribution is computed rather high terms in the multipole expansion of the
charge can be afforded.
8.17.1.5.2 The Polarizable Continuum Model, PCM
The PCM has been developed in order to describe the solvent reaction field in a
more realistic way, basically through the use of cavities of general shape, modeled on the
solute. The cavity is built as the envelope of spheres centered on solute atoms or atomic
groups (usually, hydrogen atoms are included in the same sphere as the heavy atoms they are
bonded to). The reaction field is described by means of apparent charges (solvation
charges) spread on the cavity surface, designed to reproduced the electrostatic potential
due to the polarized dielectric inside the cavity.
Such charges are used both to compute solute-solvent interactions (modifying the total energy
of the solute), and to perturb the molecular Hamiltonian through a suitable operator
(thus distorcing the solute wave-function, and affecting all the electronic properties).
The PCM operator contains both one- and two-electron terms: it is computed using
atomic integrals already present in the program, through a ``geometry matrix''
connecting different points lying on the cavity surface. It can be shown that
with this approach the SCF and RASSCF variational procedures lead to the
free energy of the given molecule in solution: this is the thermodynamic meaning
of the SCF or CI energy provided by the program. More precisely, this is the
solute-solvent electrostatic contribution to the free energy
(of course, other terms depending on solute atomic motions, like vibrational and
rotational free energies, should be included separately);
it can be used to get a good
approximation of the solvation free energy, by subtracting the SCF or CI energy
computed in vacuo, and also to compute directly energy surfaces and reaction paths
in solution. On the other hand, the solute wave-function perturbed by the
reaction field can be used to compute any electronic property in solution.
Also other quantities can be computed, namely the cavitation free energy (due the the work spent to create the cavity in the dielectric) and the dispersion-repulsion free energy: these terms affect only the total free energy of the molecule, and not its electronic distribution. They are collectively referred to as non-electrostatic contributions.
Note that two other keywords are defined for the RASSCF program: they refer to the CI root selected for the calculation of the reaction field (RFROOT), and to the possibility to perform a non-equilibrium calculation (NONEQ) when vertical electronic transitions are studied in solution. These keywords are referenced in the RASSCF section. To include the reaction field perturbation in a SCF, RASSCF, CASPT2 or RASSI calculation, another keyword must be specified (RFPERT), as explained in the respective program sections.
Complete and detailed examples of how to add a reaction field,
through the Kirkwood or the PCM model, into quantum chemical
calculations in MOLCAS is presented in section of the
examples manual. The user is encouraged to read that section for further details.
8.17.1.5.3 Input for the Kirkwood and PCM models
8.17.1.5.3.1 Files
The reaction field calculations will store the information in the following files, which will be used by the following programs
| File | Contents |
| ONEINT | One-electron integral file used to store the Pauli repulsion integrals |
| RUNFILE | Communications file. The last computed self-consistent reaction field (SCF or RASSCF) will be stored here to be used by following programs |
| GV.off | Input file for the external program ``geomview'' (see Tutorial section ``Solvent models''), for the visualization of PCM cavities |
8.17.1.5.3.2 Input
Below follows a description of the input to the reaction field utility in the GATEWAY program. The RASSCF program has its own keywords to compute reaction fields for excited states.Compulsory keywords
| Keyword | Meaning |
| RF-Input | Activate reaction field options.
|
| END Of RF-Input | This marks the end of the input to the reaction field utility. |
Optional keywords for the Kirkwood Model
| Keyword | Meaning |
| REACtion Field | This command is exclusive to the Kirkwood model. It indicates the beginning of the specification of the reaction field parameters. The subsequent line will contain the dielectric constant of the medium, the radius of the cavity in Bohrs (the cavity is always centered around the origin), and the angular quantum number of the highest multipole moment used in the expansion of the change distribution of the molecule (only charge is specified as 0, charge and dipole moments as 1, etc.). The input specified below specifies that a dielectric permitivity of 80.0 is used, that the cavity radius is 14.00 a.u., and that the expansion of the charge distribution is truncated after l=4, i.e. hexadecapole moments are the last moments included in the expansion. Optionally a fourth argument can be added giving the value of the dielectric constant of the fast component of the solvent (default value 1.0). |
RF-Input
Reaction field
80.0 14.00 4
End Of RF-Input
Sample input for a complete reaction field calculation using the Kirkwood model. The SCF computes the reaction field in a self consistent manner while the MRCI program adds the effect as a constant perturbation.
&GATEWAY
Title = HF molecule
Symmetry
X Y
Basis set
F.ANO-S...3S2P.
F 0.00000 0.00000 1.73300
End of basis
Basis set
H.ANO-S...2S.
H 0.00000 0.00000 0.00000
End of basis
Well integrals
4
1.0 5.0 6.75
1.0 3.5 7.75
1.0 2.0 9.75
1.0 1.4 11.75
RF-Input
Reaction field
80.0 4.75 4
End of RF-Input
&SEWARD
&SCF
Occupied = 3 1 1 0
&MOTRA
LumOrb
Frozen = 1 0 0 0
RFPert
&GUGA
Electrons = 8
Spin = 1
Inactive = 2 1 1 0
Active = 0 0 0 0
CiAll = 1
&MRCI
SDCI
Optional keywords for the PCM Model
| Keyword | Meaning |
| PCM-model | If no other keywords are specified, the program will execute a standard PCM calculation with water as solvent. The solvent reaction field will be included in all the programs (SCF, RASSCF, CASPT2, etc) invoked after SEWARD: note that in some cases additional keywords are required in the corresponding program sections. Some PCM parameters can be changed through the following keywords. |
| SOLVent | Used to indicate which solvent is to be simulated. The name of the requested solvent must be written in the line below this keyword. Find implemented solvents in the PCM model below this section. |
| DIELectric constant | Defines a different dielectric constant for the selected solvent; useful to describe the system at temperatures other that 298 K, or to mimic solvent mixtures. The value is read in the line below the keyword. An optional second value might be added on the same line which defines a different value for the infinite frequency dielectric constant for the selected solvent (this is used in non-equilibrium calculations; by default it is defined for each solvent at 298 K). |
| CONDuctor version | It requires a PCM calculation where the solvent is represented as a polarized conductor: this is an approximation to the dielectric model which works very well for polar solvents (i.e. dielectric constant greater than about 5), and it has some computational advantages being based on simpler equations. It can be useful in cases when the dielectric model shows some convergence problems. |
| AAREa | It is used to define the average area (in Å2) of the small elements on the cavity surface where solvation charges are placed; when larger elements are chosen, less charges are defined, what speeds up the calculation but risks to worsen the results. The default value is 0.4 Å2 (i.e. 60 charges on a sphere of radius 2 Å). The value is read in the line below the keyword. |
| R-MIn | It sets the minimum radius (in Å) of the spheres that the program adds to the atomic spheres in order to smooth the cavity surface (default 0.2 Å). For large solute, if the programs complains that too many sphere are being created, or if computational times become too high, it can be useful to enlarge this value (for example to 1 or 1.5 Å), thus reducing the number of added spheres. The value is read in the line below the keyword. |
| PAULing | It invokes the use of Pauling's radii to build the solute cavity: in this case, hydrogens get their own sphere (radius 1.2 Å). |
| SPHEre radius | It is used to provide sphere radii from input: for each sphere given
explicitly by the user, the keyword ``Sphere radius'' is required,
followed by a line containing two numbers: an integer indicating the
atom where the sphere has to be centered, and a real indicating its
radius (in Å). For example, ``Sphere radius'' followed by ``3 1.5''
indicates that a sphere of radius 1.5 Å is placed around atom #3;
``Sphere radius'' followed by ``4 2.0'' indicates that another sphere of
radius 2 Å is placed around atom #4 and so on.
|
Solvents implemented in the PCM model are
| Name | Dielectric | Name | Dielectric | Name | Dielectric | |||
| constant | constant | constant | ||||||
| water | 78.39 | dichloroethane | 10.36 | toluene | 2.38 | |||
| dimethylsulfoxide | 46.70 | quinoline | 9.03 | benzene | 2.25 | |||
| nitromethane | 38.20 | methylenchloride | 8.93 | carbontetrachloride | 2.23 | |||
| acetonitrile | 36.64 | tetrahydrofuran | 7.58 | cyclohexane | 2.02 | |||
| methanol | 32.63 | aniline | 6.89 | heptane | 1.92 | |||
| ethanol | 24.55 | chlorobenzene | 5.62 | xenon | 1.71 | |||
| acetone | 20.70 | chloroform | 4.90 | krypton | 1.52 | |||
| isoquinoline | 10.43 | ethylether | 4.34 | argon | 1.43 |
Sample input for the reaction field part (PCM model): the solvent is water, a surface element average area of 0.2 Å2 is requested.
RF-input
PCM-model
Solvent
water
AAre
0.2
End of RF-input
Sample input for a standard PCM calculation in water. The SCF and RASSCF programs compute the reaction field self consistently and add its contribution to the Hamiltonian. The RASSCF is repeated twice: first the ground state is determined, then a non-equilibrium calculation on the first excited state is performed.
&GATEWAY
Coord
4
formaldehyde
O 0.000000 0.000000 -1.241209
C 0.000000 0.000000 0.000000
H 0.000000 0.949585 0.584974
H 0.000000 -0.949585 0.584974
Basis = STO-3G
Group = C1
RF-input
PCM-model
solvent = water
End of RF-input
&SEWARD ; &SCF
&RASSCF
nActEl = 4 0 0
Symmetry = 1
Inactive = 6
Ras2 = 3
CiRoot
1 1
1
LumOrb
&RASSCF
nActEl = 4 0 0
Symmetry = 1
Inactive = 6
Ras2 = 3
CiRoot
2 2
1 2
0 1
JOBIPH
NonEq
RFRoot = 2
Again the user is recommended to read section of the
examples manual for further details.
8.17.1.6 Keywords associated to one-electron integrals
| Keyword | Meaning |
| FNMC | Request that the so-called Finite Nuclear Mass Correction exclude, by the Born–Oppenheimer approximation, be added to the one-electron Hamiltonian. |
| WELL integrals | Request computation of Pauli repulsion integrals for dielectric cavity reaction field calculations. The first line specifies the total number of primitive well integrals in the repulsion integral. Then follows a number of lines, one for each well integral, specifying the coefficient of the well integral in the linear combination of the well integrals which defines the repulsion integral, the exponent of the well integral, and the distance of the center of the Gaussian from the origin. In total three entries on each line. All entries in atomic units. If zero or a negative number is specified for the number of well integrals a standard set of 3 integrals with their position adjusted for the radius of the cavity will be used. If the distance of the center of the Gaussian from the origin is negative displacements relative to the cavity radius is assumed. |
| XFIEld integrals | Request the presence of an external electric field represented by a number of partial charges and dipoles. Optionally, polarisabilities may be specified whose induced dipoles are determined self-consistently during the SCF iteration. The first line may contain, apart from the first integer [nXF] (number of centers), up to four additional integers. The second integer [nOrd] specifies the maximum multipole order, or -1 signifying no permanent multipoles. Default is 1 (charges and dipoles). The third integer [p] specifies the type of external polarisabilities: 0 (default) no polarisabilities, 1 (isotropic), or 2 (anisotropic). The fourth integer [nFrag] specifies the number of fragments one multipole may contribute to (relevant only if polarisabilities are present). The default is 0, meaning that each permanent multipole is only excluded in the calculation of the field at its own polarisability, 1 means that one gives a fragment number to each multipole and that the static multipoles do not contribute to the polarising field within the same fragment, whereas 2 can be used in more complex situations, e.g. polymers, allowing you to specify a second fragment number so that junction atoms does not contribute to either of the neighbouring fragments. Finally, the fifth and last integer [nRead] (relevant only if Langevin dipoles are used) may be 0 or 1 (where 0 is default), specifying wheather an element number (e.g. 8 for oxygen) should be read for each multipole. In that case the default radius for that element is used to determine which Langevin grid points should be annihilated. A negative element number signifies that a particular radius should be used for that multipole, in thousands of a Bohr (-1400 meaning 1.4 Bohr). Then follows nXF lines, one for each center. On each line is first nFrag+nRead (which may equal 0) integers, specifying the fragments that the multipole should not contribute to (the first fragment is taken as the fragment that the polarisability belongs to) and the element number. Then follows the three coordinates of the center, followed by the multipoles and polarisabilities. The number of multipole entries is 0 for nOrd=-1, 1 for nOrd=0, 4 for nOrd=1, and 10 for nOrd=2. The number of polarisability entries are 0 for p=0, 1 for p=1, and 6 for p=2. The order of quadrupole moment and anisotropic polarisability entries is xx, xy, xz, yy, yz, zz. If default is used, i.e. only specifying the number of centers on the first line, each of these lines will contain 7 entries (coordinates, charge, and dipole vector). All entries are in atomic units, if not otherwise requested by the Angstrom keyword that must be placed between nXF and nOrd. All these data can be stored in a separate file whose name must be passed as an argument of the XField keyword. |
| ANGM | Supplement ONEINT for transition angular momentum calculations. Entry which specifies the angular momentum origin (in au). |
| OMQI | Supplement ONEINT for transition orbital magnetic quadrupole calculations. Entry which specifies the orbital magnetic quadrupole origin (in au). |
| AMPR | Request the computation of angular momentum product integrals. The keyword is followed by values which specifies the angular momentum origin (in au). |
| DSHD | Requests the computation of diamagnetic shielding integrals. The first
entry specifies the gauge origin. Then follows an integer
specifying the number of points at which the diamagnetic
shielding will be computed. If this entry is zero, the diamagnetic
shielding will be computed at each nucleus. If nonzero, then the
coordinates (in au) for each origin has to be supplied, one entry for each
origin.
|
| EPOT | An integer follows which represents the number of points for which the electric potential will be computed. If this number is zero, the electric field acting on each nucleus will be computed. If nonzero, then the coordinates (in au) for each point have to be supplied, one entry for each point. This keyword is mutually exclusive with EFLD and FLDG. |
| EFLD | An integer follows which represents the number of points for which the electric potential and electric field will be computed. If this number is zero, the electric field acting on each nucleus will be computed. If nonzero, then the coordinates (in au) for each point have to be supplied, one entry for each point. This keyword is mutually exclusive with EPOT and FLDG. |
| FLDG | An integer required which represents the number of points for which the electric potential, electric field and electric field gradient will be computed. If this number is zero, the electric field gradient acting on each nucleus will be computed. If nonzero, then either the coordinates (in au) for each point or labels for each atom center have to be supplied, one entry for each point. In case a label is supplied it must match one of those given previous in the input during specification of the coordinates of the atom centers. Using a label instead of a coordinate can e.g. be useful in something like a geometry optimization where the coordinate isn't known when the input is written. This keyword is mutually exclusive with EPOT and EFLD. |
| EMPC | Use point charges specified by the keyword XField when calculating the Orbital-Free Embedding potential. |
| RF-Input | Specification of reaction field parameters, consult the reaction field section of this manual. |
8.17.1.7 Keywords associated with nuclear charge distribution models
Input parameters associated with different models of the nuclear charge distribution. The default is to use a point charge representation.| Keyword | Meaning |
| FINIte | Request a finite center representation of the nuclei by a single exponent s-type Gaussian. |
| MGAUSsian | Request a finite center representation of the nuclei by a modified Gaussian. |
8.17.1.8 The Saddle method for transition state optimization
The Saddle method [51] is a method to locate transition states (TS). The method, in practice, can be viewed as a series of constrained optimization along the reaction path, which connects two starting structure (could be the reactants and products of a reaction), to locate the region of the TS and a subsequent unconstrained optimization to locate the TS. The only data needed for the procedure are the energies and coordinates of the two structures. Note that this option will overwrite the coordinates which have already been specified with the normal input of the molecular geometry. However, this does not make that input section redundant and should always be included.
| Keyword | Meaning |
| RP-Coordinates | This activates the Saddle method for TS geometry optimization.
The line is followed by an integer specifying the number of symmetry unique coordinates to be specified. This
is followed by two sets of input - one line with the energy and then the Cartesian coordinates in bohr - for
each of the two starting structures of the Saddle method. Note that the order of the coordinates must always
match the order specified with the conventional input of the coordinates of the molecular system.
Alternatively, two lines with the filenames containing the coordinates of reactants and products, respectively,
(in XYZ format) can be given.
|
| NOALign | By default, the two starting structures are aligned to minimize the root mean square distance (RMSD) between them,
in particular, the first structure is moved and the second structure remains fixed.
If this keyword is given, the starting structures are used as given.
|
| ALIGn only | The two starting structures are aligned, but nothing more is done.
An input block for seward is still needed, but no integrals are computed.
|
| WEIGhts | Relative weights of each atom to use for the alignment and for the calculations of the
``distance'' between structures. The possibilities are:
MASS. This is the default. Each atom is given a weight proportional to its mass. Equivalent to mass-weighted coordinates. EQUAL. All atoms have an equal weight. HEAVY. Only heavy atoms are considered, with equal weights. Hydrogens are given zero weight. A list of N numbers can also be provided, and they will be used as weights for the N symmetry-unique atoms. For example:
will align only atoms 7–12 out of 16. Note that, in any case, weights of 0 are likely to cause problems with constraints, and they will be increased automatically.
|
| SADDle | Step size reduction for each macro iteration of the saddle method.
The value is given in weighted coordinates, divided by the square root of the total weight
(see the WEIGHTS keyword).
Default value is 0.1 au.
|
8.17.1.9 Geometry optimization using constrained internal coordinates
These keyword are used together with the geo to optimize the relative position of two or more rigid fragments. The starting geometry can either be defined by supplying an xyz-file for each fragment using the keyword coord or by placing a file named $Project.zmt in a directory named $Project.GEO. The z-matrix should be in the following format:
H
O 0.982011 0 1
H 0.982013 0 104.959565 0 2 1
H 1.933697 1 107.655494 1 114.496053 1 2 3 1
O 0.988177 0 173.057942 1 -56.200750 1 4 2 3
H 0.979890 0 104.714572 0 179.879745 1 5 4 2
where the three columns of real numbers are internal coordinates, and the last three columns of integers indicate which other atoms that are used to define the coordinate. The type of coordinates from left to right are bond distances, bond angles and dihedral angels, both for the coordinates and the link. The column of integers just to the right of each coordinate indicate if this coordinate should be optimized or not (1 = optimize, 0 = do not optimize).
There are also two utility-keywords used to create a z-matrix or to write out a constraint-definition for slapaf and keywords to rotate and translate fragments. (See documentation for GEO for more details)
| Keyword | Meaning |
| HYPER | This keyword is used to specify that a geometry optimization with constrained
internal coordinates shall be performed later, a z-matrix and a set of
displaced geometries are therefore constructed. The keyword should be followed by three
real numbers defining the maximum displacement for each coordinate type.
The order from left to right is bond distances, bond angles and dihedral angles.
To use default values for the parameters the mutually exclusive keyword
geo should be entered instead.
|
| GEO | This keyword is used to specify that a geometry optimization with constrained
internal coordinates shall be performed later, a z-matrix and a set of displaced
geometries are therefore constructed. Default values of 0.15 Å, 2.5 degrees,
and 2.5 degrees are used for the maximum displacement of bond distances, bond
angles and dihedral angles respectively. To enter other values for the parameters
the mutually exclusive keyword hyper should be used.
|
| OPTH | This keyword is used to define the specific details of the optimization algorithm used
for the geometry optimization in constrained internal coordinates.
This keyword should be followed by two to three lines of parameter. The first line should
contain an integer indicating optimization type (1 = steepest descent, 2 = a mix of
steepest descent and Newton's method, and 3 = Newton's method). The second line
should contain a real number defining a step factor.
This number is multiplied with the gradient to obtain the step length.
For optimization type 2 a third line containing a real number that defines a gradient limit
should be entered. This limit determines how large the gradient must be for the steepest
descent algorithm to be used. When the gradient is smaller than this limit Newton's method
is used instead.
|
| OLDZ | This keyword is used both to start a new calculation from a user-defined z-matrix and
to restart calculations. When using the keyword for a new calculation a directory
$Project.GEO must exist and contain a file called $Project.zmt with a z-matrix in
the format defined above. The directory must not contain any files with the suffix .info
when performing a fresh calculation since these files contain restart information.
|
| ZONLY | This keyword is used to construct a z-matrix from a set of xyz-files (fragments)
and store it in the directory $Project.GEO. The optimization parameters
of the resulting z-matrix are set so that only coordinates linking fragments are
set to 1 (= optimize coordinate).
|
| ZCONS | This keyword is used to define constraints from a set of xyz-files (fragments)
on a form that could be supplied to the
slapaf in order to keep the fragments rigid. The resulting constraints-file
is named $Project.cns and stored in the directory $Project.GEO. The
atom-numbers in this constraint-file will not match those of your original xyz-file and
should not be used together with this. Instead a new xyz-file named cons.xyz is created
and placed into the directory $Project.GEO, this has the proper numbering to use together with the constraints.
|
| ORIGIN | This keyword is used to translate and rotate a set of xyz-files. The keyword must be entered
before the xyz-files is entered with coord.
The keyword should be followed by two lines for each fragment in the input.
The first row should contain 3 real numbers defining a translation (x, y, z),
the second row should contain 9 numbers defining a rotation (row1, row2, row3 of
rotation matrix). The keyword origin is mutually exclusive with the keyword frgm
which is an alternative way to express the same rotations and translations.
|
| FRGM | This keyword is used together with the keywords rot and trans to define
rotation and translation of a specific fragment. Frgm defines an active fragment (each xyz-file is considered a fragment, the files are numbered based on
order of appearance in the input from top to bottom). The keyword must be entered before the xyz-file it is supposed to modify is
entered with coord. Each occurence of
frgm should be followed by either one of or both of the keywords rot and trans
to define rotation and translation. This keyword is mutually exclusive with the keyword orgin
|
| ROT | This keyword should be followed by nine real numbers defining the rotation for the fragment defined by
the preceeding frgm. The numbers should be the nine elements of a rotation matrix
listed with one full row at the time.
|
| TRANS | This keyword should be followed by three real numbers defining the translation for the fragment defined
by the preceeding frgm. The numbers should be the x, y and z coordinates of the translation
in that order.
|
Example of an input:
&GATEWAY
Title
Water Dimer
frgm=2
trans=3.0 0.0 0.0
Coord=water_monomer.xyz
Coord=water_monomer.xyz
Group=c1
basis=cc-pVTZ
hyper
0.2 3.0 3.0
opth
3
15.0d0
In this example a water dimer is constructed from a single monomer by translating it 3.0 Åwith the keyword trans. An optimization in constrained internal coordinates using newtons method with a step-factor of 15.0d0 are prepared for. For more details on these optimization see the manual entry for the module geo.
8.17.1.10 QM/MM calculations with MOLCAS/Gromacs
The following keywords apply to QM/MM calculations performed with the MOLCAS/GROMACS interface (see section for more details).
| Keyword | Meaning |
| GROMacs | Requests that the definition of the full QM+MM system should be imported from GROMACS. The keyword should be followed by one of the options SIMPLE or CASTMM on the next line. In the case of SIMPLE, all MM atoms defined in the GROMACS input will be treated as outer MM atoms in MOLCAS. This means, for example, that in a geometry optimization, their positions will be updated using microiterations rather than the conventional optimization scheme. Conversely, CASTMM requests that certain MM atoms should be treated as inner MM atoms in MOLCAS. Their positions will be updated with the same scheme as used for the QM atoms. The CASTMM option should be followed by two additional input lines, the first one containing the number of MM atoms to convert from outer to inner type, and the second containing a list of those atoms (using their corresponding GROMACS indices).
|
| LINKatoms | Defines link atoms for use with the Morokuma updating scheme. The desired number of link atoms should be given as an integer on the next line. This should be followed by additional input lines, one for each link atom to be defined. Each definition should be of the form ILA, IQM, IMM, SCALE, where ILA, IQM and IMM are the GROMACS indices of the link atom and the corresponding QM and MM frontier atoms, respectively. SCALE is the scaling factor to be used in the Morokuma scheme. Note that each link atom must be defined as a QM atom in the GROMACS input. In addition, the frontier MM atom must be an inner MM atom specified as discussed above.
|
Next: 8.18 genano Up: 8. Programs Previous: 8.16 ffpt

